HyLiTE documentation, classes and code¶
This page gives a detailed account of the structure of HyLiTE’s code. If you are looking for HyLiTE’s basic usage, please see the HyLiTE manual page instead.
HyLiTE’s code is contained in a module called hylite.
The following figure describes the organisation of hylite’s classes in an UML fashion:
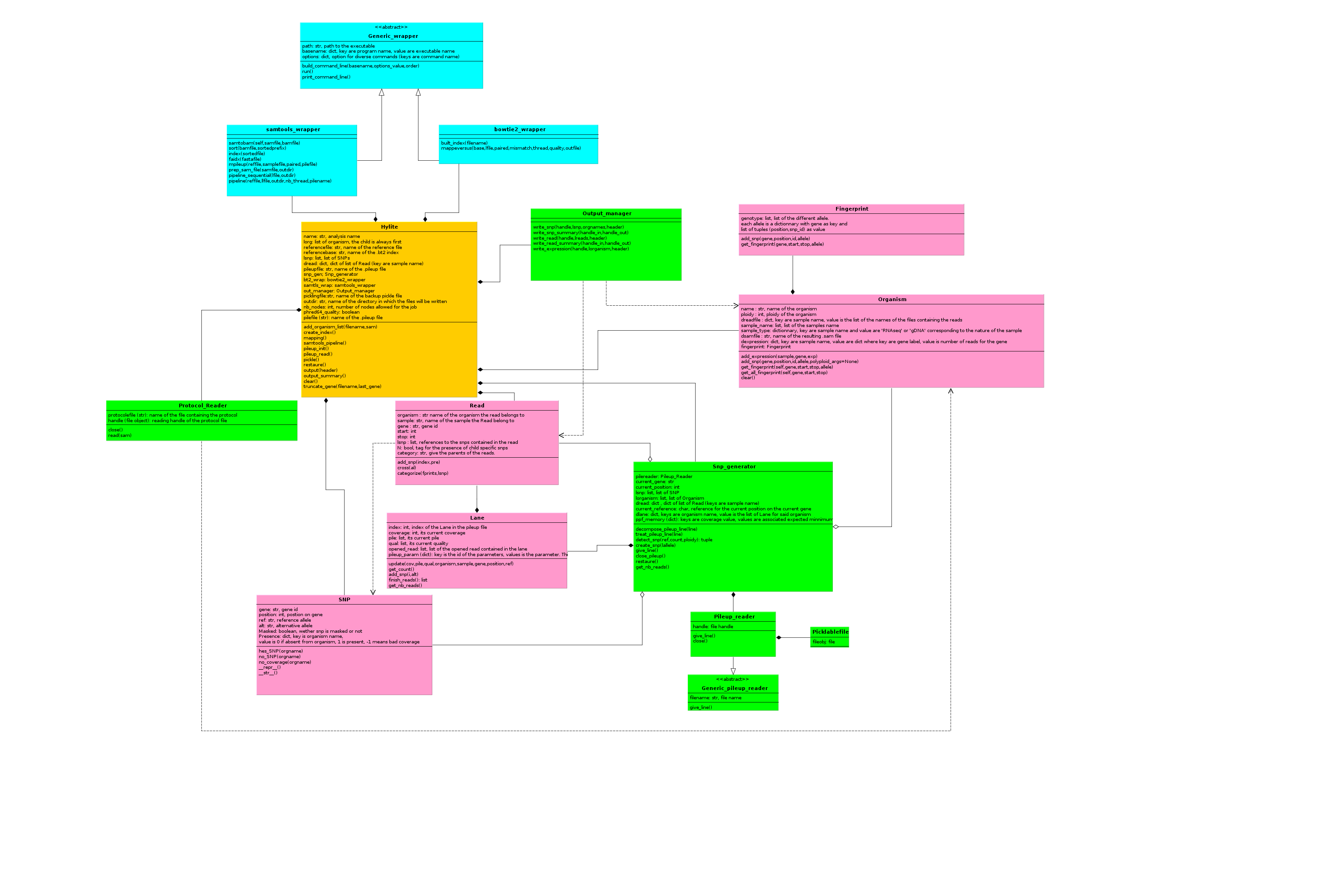
Hylite class¶
-
class
hylite.Hylite.Hylite(name, outdir, nb_threads, phred64_quality)[source]¶ Bases:
objectMain class of HyLiTE. Coordinate all the different analysis and ensure proper output of the results
- Attributes:
lorg (list): list of organism. THE CHILD ORGANISM IS ALWAYS THE FIRST OF THE LIST
referencefile (str): name of the .fasta file containing the reference sequences/gene model for the analysis
referencebase (str): name of the .bt2 index
lsnp (list): list of SNP objects, modeling the repartition of snps between different organisms
dread (dict):dict (keys are sample name) of list of the child’s Read, containing where they align on the reference model, their snp content, and their parental origin
snp_gen (Snp_Generator)
bt2_wrap (bowtie2_wrapper)
samtls_wrap (samtools_wrapper)
out_manager (Output_manager)
picklingfile (str): name of the backup pickle file
name (str): analysis name
outdir (str): name of the directory in which the files will be written
nb_nodes (int): number of nodes allowed for the job
phred64_quality (boolean): boolean set to True if the quality of the readfiles is in phred64 quality
pilefile (str): name of the .pileup file
last_gene (str): id of the last written gene before the last pickle
-
add_organism_list(filename, sam)[source]¶ Add the list of organism to HyLiTE from a file given as argument
- Args:
filename (str): the name of the file containing the organism informations
sam (bool): set to True if the protocol file contains in fact the .sam files
- FORMAT of the file:
without header
separated by ‘ ‘
one line per sample per organism
SAMPLE_TYPE is ‘RNAseq’ or ‘gDNA’ (if other, DEFAULT_SAMPLE_TYPE will be put instead)
THE CHILD MUST BE THE FIRST ORGANISM
ORGANISM_NAME PLOIDY SAMPLE_NAME SAMPLE_TYPE READ_FILE.fastq
-
output(header)[source]¶ Output the different results of HyLiTE
- Args:
header (bool): True if the header of the file must be written
-
pileup_read()[source]¶ This function execute the main algorithm of HyLiTE: it will call the SNPs for each position and then assign a category to each of the child’s read this function also check the need for pickling
-
restaure()[source]¶ This function is used to start the SNP calling again from the last gene completed. It is used after an unpickling of the Hylite instance
-
samtools_pipeline()[source]¶ Proceed to the treatment of the different .sam file in order to obtain the .pileup file
-
set_referencefile(referencefile)[source]¶ Set the reference file in the current HyLiTE instance as well as in the outputmanager. The output manager needs it to know which genes to write in the output files
-
truncate_gene(filename, last_gene)[source]¶ This function allow to truncate a result file after a specific gene. It is useful when unpickling an ancient HyLiTE instance. (desynchronisation between the writing of results and pickling)
- Args:
filename (str): name of the file to truncate
last_gene (str): id of the last gene before the pickling took place
Read class¶
-
class
hylite.Read.Read(organism, sample, gene, start)[source]¶ Bases:
objectUsed to represent a read.
- Attributes:
organism (str): name of the organism the read belongs to
sample (str): name of the sample the Read belong to
gene (str): gene id
start (int): starting position of the read on the gene
stop (int): stopping position of the read on the gene
lsnp (list): references to the snps contained in the read
N (bool): tag for the presence of child specific snps
category (str): give the parents of the reads.
-
add_snp(index, pre)[source]¶ Add a snp to the read
- Args:
index (int): the index of a snp
pre (int): presence of the snp at this position in the read
-
categorize(fprints, lsnp)[source]¶ Compare each fingerprint to its own and assign a category to the read. The finger prints are in fact list of snp index.
- Args:
fprints (dict): dictionnary (key is organism name) of list of list of tuple (snp index,presence) for each allele of each parent between start and stop
lsnp (list): the list of all snps
- Note:
Fingerprints don’t contain masked snps
- Category notation:
If the read contain at least one snp belonging only to the child, a tag N is activated
Belonging to any specific parental category is stocked as a string identifier
Fingerprint class¶
-
class
hylite.Fingerprint.Fingerprint(ploidy)[source]¶ Bases:
objectClass used to determine the possible snp distribution of an organism in a gene between two given positions (called fingerprint)
- Attributes:
genotype (list)
-
add_snp(gene, position, id, presence, allele)[source]¶ Add the given snp index (and presence -1/0/1) at given position of gene on the given allele (total number of allele = ploidy of the organism)
- Args:
gene (str): name of the gene containing the snp
position (int): position of the snp on the gene
id (int): index of the snp in the list of snp of Hylite
presence (int): can take the values -1 (bad coverage) or 0 (absence) or 1 (presence)
allele (int): index of the allele to which we should add the snp
-
get_fingerprint(gene, start, stop, allele)[source]¶ Return the list of the tuple (snps_index,presence) present between start and stop on the given gene on the given allele
- Args:
gene (str): name of the gene containing the snp
start (int): starting position for the fingerprint
stop (int): stopping position for the fingerprint
allele (int): index of the concerned allele
- Returns:
list. actually a list of tuple (snp_index,presence)
bowtie2_wrapper class¶
-
class
hylite.bowtie2_wrapper.bowtie2_wrapper[source]¶ Bases:
hylite.Generic_wrapper.Generic_wrapperThis class is a wrapper for bowtie2 inheriting from Generic_wrapper
- Attributes:
commandline (str): commandline used
-
build_index(reffile, outname)[source]¶ Function used to build the .bt2 index from a reffile fasta file
- Args:
reffile (str): a reference file name (file is fasta)
outname (str): the name of the .bt2 base to create
-
mappe_versus(base, lfile, paired, mismatch, thread, quality64, outfile)[source]¶ Function used to map reads against a base using bowtie2.
- Args:
base (str): the name of the .bt2 base
lfile (list): a list of the name of the file containing the reads
paired (bool): a boolean set to True of the data are paired end reads
mismatch (bool): a boolean set to True of you want to allow mismatch in the seed
thread(int): an int specifying the number of thread to use (1 is authorized)
quality64 (bool): a boolean set to True if the quality of the reads is in phred64 format
outfile (str): a string specifying the name of the .sam out file to write
Generic_Pileup_Reader class¶
Generic_wrapper class¶
-
class
hylite.Generic_wrapper.Generic_wrapper(path, basename, options)[source]¶ Bases:
objectThis abstract class represent a wrapper
- Attributes:
path (str): the path of the software
basename (dict): keys are of the software function, values are the corresponding command
option (dict): keys are the options name, values are their usage in the command line
-
build_command_line(basename, options_value, order)[source]¶ Build the command line that the run method will use.
- Args:
basename (str):the name of command he want to use
options_value (dict): a dictionnary containing the options he want to use: the key are the option name, the value are the one associated with the option
order (list): a list containing the ordered name of options (order of appearance in the command line)
- Example:
If we consider a class grep_wrapper inheriting from this class. basename would comprise only {‘grep’:’grep’} and option could be something like {‘inverse’:’-v’,’count’:’-c’,’pattern’:’’,’input’:’’}. To build a grep command, the user would have to provide the basename ‘grep’ and a dictionnary of option as follow: DICT RESULTING GREP COMMAND {‘pattern’:’”^>”’,’input’:’file1.fasta’} grep “^>” file1.fasta {‘count’:True,’pattern’:’”^>”’,’input’:’file1.fasta’} grep -c “^>” file1.fasta {‘inverse’:False,’count’:True,’pattern’:’”^>”’,’input’:’file1.fasta’} grep -c “^>” file1.fasta
-
print_command_line()[source]¶ Print the current command line
- Raises:
AttributeError Exception. if you haven’t defined a command line before
-
run()[source]¶ Execute the commandline NB: as this is an abstract class, it does not contain an attribute command line and will thus raise an error if directly executed. Any inheriting class have to define a commandline attribute
- Returns:
Popen. Popen of the PIPED command
- Raises:
AttributeError Exception. if you haven’t defined a command line before
Lane class¶
-
class
hylite.Lane.Lane(index)[source]¶ Bases:
objectThis class is used to represent a lane in a pileup file (a lane is comprised of three column: coverage, pile, quality).
- Attributes:
index (int): its index in the pileup file
coverage (int): its current coverage
pile (str): its current pile
qual (list): its current quality
opened_read (list): a list of the opened read contained in the lane
pileup_param (dict): key is the id of the parameters, values is the parameter. This is mostly the character .pileup use to represent data
-
add_snp(i, alt)[source]¶ Add the snp to the reads
- Args:
i (int): the index of the spn
alt (str): the alt of the snp
-
finish_reads()[source]¶ Finish the reads properly and return them
- Returns:
list. a list of the finished reads
-
get_count()[source]¶ - Returns:
dict. a dictionnary containing the count of each letter (key are letter, value is count)
-
update(cov, pile, qual, organism, sample, gene, position, ref)[source]¶ Update the Lane with this information
- Args:
cov (int): a coverage
pile (str): a pile
qual (str): a quality str
organism (str): an organism name
sample (str): a sample name
gene (str): a gene name
position (int): a position
ref (str): the reference for the position
Update the tags of its opened reads according to their content and the list of detected alleles at this position
- Args:
allele (list): list of the detected alleles at this position
!!!This is not made for more than diploid yet
Organism class¶
-
class
hylite.Organism.Organism(name, ploidy, dreadfile, sample_name, sample_type)[source]¶ Bases:
objectThis class represent an organism (parent or child) in our analysis
- Its attributes are:
name : str, name of the organism
ploidy : int, ploidy of the organism
dreadfile : dict, key are sample name, value is the list of the names of the files containing the reads
sample_name: list, list of the samples name
sample_type: dictionnary, key are sample name and value are ‘RNAseq’ or ‘gDNA’ corresponding to the nature of the sample
dsamfile : str, name of the resulting .sam file
dexpression: dict, key are sample name, value are dict where key are gene label, value is number of reads for the gene
fingerprint: Fingerprint
-
add_expression(sample, gene, exp)[source]¶ Increment the count of a given gene from a given sample
- Args:
sample (str): a sample name
gene (str): a gene name
exp (int): the number of count
-
add_snp(gene, position, snp_index, presence, polyploid_args=None)[source]¶ - Args:
gene (str): a gene name
position (int): the position of the snp on the gene
snp_index (int): index of the snp in the hylite list
presence (list): list of int; give its presence (-1/0/1) in each gene copy
- Kwargs:
polyploid_args: optional argument for polyploids #actually not used… but whatever, let’s keep it
Output_manager class¶
-
class
hylite.Output_manager.Output_manager[source]¶ Bases:
objectThis class manages the outputs of HyLiTE
-
get_all_genes()[source]¶ Function finding all genes in the reference file and saving them in a class variable. The name of the genes is found based on the same method as BioPython FastaIterator: the gene name is the first word of the fasta defline.
-
precompute_expression_data_towrite(lorganism, header)[source]¶ Precompute the expression data to be written in a file for the genes in each organism and each RNA-seq sample
- Args:
handle (file object): a handle to write in
lorganism (list): a list of organisms
header (bool): True if the header of the file must be written
-
simplify_category(cat)[source]¶ - Args:
cat (str): a read category
- Returns:
(str): the read category, simplified
-
write_complete_expression_file(outdir, name)[source]¶ Write the precomputed expression data in a file. All genes present in the reference file will be present in the expression file
- Args:
outdir (str): the directory to write the file in
name (str): the name of the file
-
write_read(handle, lreads, header, lorg)[source]¶ Write the reads of a sample in a specified handle
- Args:
handle (file object): a handle to write in
lreads (list): a list of reads
header (bool): True if the header of the file must be written
-
write_read_summary(handle_in, handle_out, lorg)[source]¶ Write a summary of the reads of each genes in a sample in a specified handle
- Args:
handle_in (file): handle of the file containing all the reads
handle_out (file): handle of the file to write the read summary
lorg (list): list of organism names (the first is the child)
-
write_run_summary(handle_out, lhandle_in_read, handle_in_snp, lorg)[source]¶ Writes a summary of the run’s result
- Takes:
handle_out (file object): a handle to write in
lhandle_in_read (list): list of (file object): handle to the read summary file
handle_in_snp (file_object): a handle to the snp summary file
lorg (list): list of organism names (the first is the child)
-
Parameters class¶
Picklablefile class¶
Pileup_Reader class¶
-
class
hylite.Pileup_Reader.Pileup_Reader(filename)[source]¶ Bases:
hylite.Generic_Pileup_Reader.Generic_Pileup_ReaderThis class is used to access the information contained in a .pileup file containing all parents
- Attributes:
handle (Picklablefile)
Protocol_Reader class¶
-
class
hylite.Protocol_Reader.Protocol_Reader(filename)[source]¶ Bases:
objectClass designed to read a file containing information about the organisms, samples and files of the HyLiTE analysis
- Attributes:
protocolefile (str): name of the file containing the protocol
handle (file object): reading handle of the protocol file
-
read(sam)[source]¶ Read the protocol file
- Args:
sam (bool): a boolean set to True if the protocol file contains .sam file and not reads file
- Returns:
list. the lis of the organism included in the HyLiTE analysis
- FORMAT of the file:
without header
separated by ‘ ‘
one line per sample per organism
SAMPLE_TYPE is ‘RNAseq’ or ‘gDNA’ (if other, DEFAULT_SAMPLE_TYPE will be put instead)
THE CHILD MUST BE THE FIRST ORGANISM
ORGANISM_NAME PLOIDY SAMPLE_NAME SAMPLE_TYPE READ_FILE.fastq/ALIGNMENT_FILE.sam
samtools_wrapper class¶
-
class
hylite.samtools_wrapper.samtools_wrapper[source]¶ Bases:
hylite.Generic_wrapper.Generic_wrapperA wrapper for samtools
-
faidx(fastafile)[source]¶ Index the fasta file so it can be used by mpileup/sort/index as a reference
- Args:
fastafile (str): a fasta file name
-
index(sortedfile)[source]¶ Index a sorted .bam file
- Args:
sortedfile (str): name of a sorted .bam file
-
mpileup(reffile, samplefile, paired, pilefile)[source]¶ Make the pileup file containing all SNP and reads data.
- Args:
reffile (str): the name of the indexed fasta file used as reference
samplefile (str): the name of a file containing the list of the sorted .bam files to pileup (one file by line, grouped by organism, child always first)
paired (bool): a boolean set to True if at least one organism has paired end reads
pilefile (str): the name of the pileup file to write
-
pipeline(reffile, llfile, outdir, nb_thread, pilename)[source]¶ Uses the allowed threads to convert/sort/index the different sam files. Execute then the mpileup command to create the pileup.
- Args:
reffile (str): the name of the reference file (.fasta)
llfile (list): a list of list of file (ordered by organism and sample)
outdir (str): the path of the directory where the files will be written
nb_thread (int): the number of threads allowed
pilename (str): the name of the pileupfile to create
- Returns:
str. name of the .pileup file
-
pipeline_sequential(lfile, outdir)[source]¶ Treat sequentially a list of files
- Args:
lfile (list): a list of .sam filenames (ordered by organism and sample)
outdir (str): the path of the directory where the files will be written
-
prep_sam_file(samfile, outdir)[source]¶ Convert to .bam, sort and index the file
- Args:
samfile (str): a .sam file name
outdir (str): the path of the directory where the files will be written
-
Snp_Generator class¶
-
class
hylite.Snp_Generator.Snp_Generator(pileupfile, lsnp, lorganism, dread)[source]¶ Bases:
objectThis class contains the main loops of HyLiTE algorithm. It perform SNP call, expression count and read categorization on a single read of the pileup file.
- Attributes:
pilereader (Pileup_Reader)
current_gene (str)
current_position (int)
lsnp (list): list of SNP
lorganism (list): list of Organism
dread (dict): dict of list of Read (keys are sample name)
current_reference (str): reference for the current position on the current gene
dlane (dict): keys are organism name, value is the list of Lane for said organism
ppf_memory (dict): keys are coverage value, values are associated expected minnimum count for non-error bases
-
create_snp(lallele)[source]¶ Create the necessary number of SNP for this location (possibly 0).
- Args:
lallele (list): a list containing a list of the allele for each organism (empty if the coverage is bad).
- Returns:
list. a list of the index of the new SNPs in self.lsnp
-
decompose_pileup_line(line)[source]¶ Update the different Lanes with the current line information
- Args:
line (str): a line coming from a pileup file
-
detect_snp(ref, count, ploidy)[source]¶ Call the SNPs
- Args:
ref (str): the reference letter,
count (dict): a dictionary containing the count for each letter
ploidy (int): the ploidy of the concerned organism
- Returns:
list. a list (of length equal to the ploidy) containing the possible allele (including reference) at this location
-
treat_pileup_line(line)[source]¶ Decompose the line; call SNPs; categorize and stock the reads of the different samples
- Args:
line (str): a line of the .pileup file
Update the tags of the opened reads of one parent according to their content and the list of detected alleles at this position
- Args:
allele (list): list of the detected alleles at this position
org_name (str): name of the organism to update
- Returns:
list. list of detected alleles, ordered, accepting doublons
!!!This is not made for more than diploid yet
SNP class¶
-
class
hylite.SNP.SNP(gene, position, ref, alt)[source]¶ Bases:
objectThis class is used to model a SNP, including its position on the reference genome and the presence/absence/coverage of a SNP in multiple organisms
- Attributes:
gene (str): the gene containing the snp
position (int): the position of the snp on the gene
ref (str): the reference allele
alt (str): the alternative allele
masked (bool): a boolean indicating if at least one organism has a bad coverage at the snp position
presence (dict): key is organism name, value a tuple containing is 1 if SNP is present on a gene copy, 0 if absent, -1 if the coverage is bad
-
has_SNP(orgname, i)[source]¶ Update the SNP with its presence in an organism
- Args:
orgname (str): name of an organism
i (int): gene copy the snp is present on, OR -1 if it is the child